
杯狀細胞作為腸道黏液屏障的核心細胞,其黏液分泌依賴未折疊蛋白反應(UPR)傳感器IRE1β的精準調控——IRE1β通過RIDD機制調控MUC2黏液的合成與分泌[1,2],但其活性如何適配黏液折疊負荷一直是未解之謎。AGR2作為杯狀細胞特異性二硫鍵異構酶,其功能異常與炎癥性腸病(IBD)密切相關,但此前未發現其與UPR通路的關聯[3,4]。本研究通過高通量實驗技術與分子機制驗證,揭示AGR2作為IRE1β的特異性調控因子,通過破壞其dimer形成抑制核酸酶活性,且疾病相關突變會喪失該調控功能。其中,I.DOT非接觸式納升級移液系統為高通量RT-qPCR定量提供了精準支撐,助力快速驗證分子互作與信號通路變化,為理解杯狀細胞穩態失衡與IBD的關聯提供了全新視角。
構建IRE1α敲除(ERN1?/?)的Calu-1(非杯狀細胞)和LS174T(杯狀細胞樣)細胞系(CRISPR-Cas9技術,Figure 1B),通過慢病毒轉導建立多西環素誘導的IRE1β-FLAG表達系統,以及AGR2及其突變體(C81S、H117Y等)的穩定表達細胞系。選取野生型與Agr2?/?小鼠結腸組織用于體內驗證(Figure 2E)。

圖1. IRE1β活性在杯狀細胞中過表達時減弱。A圖,RT-qPCR分析ERN2IRE1β在人細胞系中的轉錄表達。對培養物進行三次取樣,發現ERN2的表達與LS174T親本細胞中的表達有關。用于進一步研究的細胞系示意圖。首先,利用CRISPR/Cas9建立了LS174T和CALU-1親本的ERN1-/-克隆。然后,用Teton模塊轉導ERN1-/-細胞表達多西環素調控的β-FLAG。C圖顯示了過度表達IRE1β-FLAG的培養物的表型。左圖顯示未經處理的培養物,中圖顯示接受1μg/ml多西環素作用72小時的培養物(從而表達IRE1β-FLAG),右圖顯示接受1μg/ml多西環素和1μM IRE1核酸內切酶抑制劑4μ8C的培養物。代表了三個獨立的實驗。D Western印跡驗證(C)中使用的細胞系中的轉基因表達。收集轉基因誘導24小時后剩余的貼壁細胞,用免疫印跡法檢測FLAG-IRE1β的表達。以微管蛋白作為載藥對照。感染的多重性(MOI)表示添加的病毒顆粒的理論數量。E,F RT-qPCR分析轉基因誘導后24小時XBP1s/T(E)和BLOC1S1轉錄水平(F)。代表三個獨立的實驗,每個條件有三個重復。(E)下圖顯示XBP1在常規PCR檢測的同一樣本中的剪接。快速遷移帶為剪接XBP1轉錄本,慢遷移帶為XBP1非剪接轉錄本。

圖2. 粘蛋白伴侶agr2是ire1β的杯狀細胞特異性相互作用分子。A圖,B和C的MS分析樣本中標志標記IRE1的轉基因表達和免疫沉淀的成功驗證用抗FLAG檢測IRE1-FLAG的表達,并以肌動蛋白作為負載對照。B圖為用于進一步研究的細胞系示意圖。首先,利用CRISPR/Cas9建立了LS174T和CALU-1親本的ERN1-/-克隆。然后,用Teton模塊轉導ERN1-/-細胞表達多西環素調控的β-FLAG。C圖顯示了過度表達IRE1β-FLAG的培養物的表型。左圖顯示未經處理的培養物,中圖顯示接受1μg/ml多西環素作用72小時的培養物(從而表達IRE1β-FLAG),右圖顯示接受1μg/ml多西環素和1μM IRE1核酸內切酶抑制劑4μ8C的培養物。代表了三個獨立的實驗。D圖的Western印跡驗證(C)中使用的細胞系中的轉基因表達。收集轉基因誘導24小時后剩余的貼壁細胞,用免疫印跡法檢測FLAG-IRE1β的表達。以微管蛋白作為載藥對照。感染的多重性(MOI)表示添加的病毒顆粒的理論數量。E,F RT-qPCR分析轉基因誘導后24小時XBP1s/T(E)和BLOC1S1轉錄水平(F)。代表三個獨立的實驗,每個條件有三個重復。(E)下圖顯示XBP1在常規PCR檢測的同一樣本中的剪接。快速遷移帶為剪接XBP1轉錄本,慢遷移帶為XBP1非剪接轉錄本。
通過親和純化質譜(AP-MS)篩選IRE1β相互作用蛋白(Figure 2A-C),Co-IP(含Avi-tag/FLAG標簽系統)驗證AGR2與IRE1β的特異性結合(Figure 2D-E、4E-F);凝膠過濾層析分析IRE1β的寡聚化狀態(Figure 4B-C);RT-qPCR定量XBP1剪接效率與RIDD靶基因(BLOC1S1)表達(重點:采用I.DOT完成384孔板的反應混合液制備與樣品轉移,保障高通量定量的精準性;AnnexinV/活死染色定量細胞死亡(Figure3C、5E);AlphaFold2進行AGR2-IRE1β相互作用結構建模。

圖3. 共表達可抑制IRE1β的核酸內切酶活性。A圖用1μg/ml多西環素誘導轉基因24小時后,進行XBP1S/T和BLOC1S1轉錄水平的RT-qPCR分析。下圖顯示了XBP1在相同樣本中的剪接情況,這些樣本通過常規的聚合酶鏈式反應檢測。代表三個獨立的實驗,每個條件有三個重復。B圖片顯示了過度表達IRE1agr2-FLAG和不表達β的培養物的表型。左側面板顯示未經處理的培養物,中間面板顯示接受1μg/ml多西環素72小時的培養物,右側面板中的培養物接受1μg/ml多西環素和1μM 4μ8C。代表了三個獨立的實驗。48小時后,在加入和不加入外源β的情況下,對高表達IRE1AGR2-FLAG的細胞死亡進行定量。培養皿中所有細胞經AnnexinV和Live/Dead染色后進行流式細胞儀分析。所有單陽性和雙陽性細胞均為死亡細胞。代表2個獨立實驗,每個條件有三個重復。D分析IRE1、β-FLAG和AGR2在用于C的細胞系中的表達。僅收集仍附著在培養皿中的細胞,并用抗FLAG和抗AGR2來檢測IRE1β和AGR2的表達。以微管蛋白作為載藥對照。在LS174TERN1-/-IRE1FLAG-DOX細胞中驗證agr2基因敲除效率。NTC是siRNA的非靶向控制池,# 1和 # 3是siRNA的靶向AGR2。4μ8C或DMSO(載體)部分擊倒和/或處理后XBP1剪接的變化。剪接顯示為NTC/Vehicle處理的細胞的log2倍變化。E和F代表三個獨立的實驗,每個條件有三個重復。G蛋白印跡確證(E)和(F)。72小時后提取蛋白質,檢測XBP1s、AGR2和微管蛋白的表達。
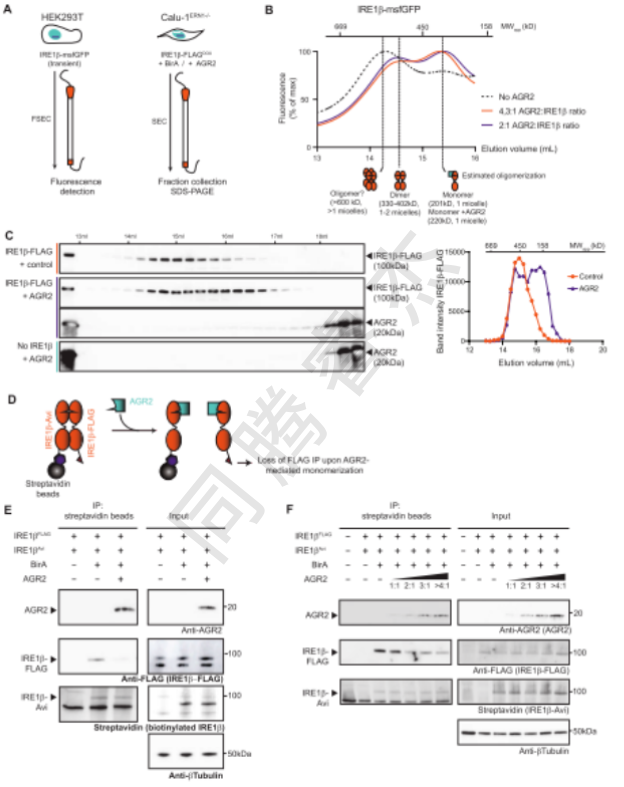
圖4. Agr2通過破壞ire1β二聚體來阻斷β的活性。A圖為B和C的凝膠過濾實驗的示意圖概述B在HEK293T裂解物的洗脫過程中測量到的msfGFP熒光,該裂解物在沒有agr2(黑點跡線)或存在agr2(橙色和紫色痕跡表示不同比例的轉基因agr2:ire1ββ)的情況下高表達ir1 DNA。最高標度代表大致的洗脫圖譜和蛋白質標準的預期分子量。底圖顯示了基于蛋白質標準和先前獲得的洗脫圖譜(灰色)的預期齊聚狀態。實驗進行了一次。在沒有或存在額外β表達的情況下,從CALU-1ERN1-/IRE1AGR1FLAG-DOX細胞的蛋白裂解物凝膠過濾后收集的組分中c IRE1AGR2-FLAG的表達。折線圖顯示了凝膠中的帶強度的量化。代表了兩個獨立的實驗。E和F中競爭IP實驗的D示意圖。IRE1β用等摩爾量的Avittag或FLAG標記表示。在被Bira生物素化后,如果已經形成二聚體,則在鏈霉親和素IP之后將同時檢測到avi標簽和FLAG標簽。如果添加另一種蛋白質(例如AGR2)會阻止這一過程,預計會失去信號。E競爭IP顯示AGR2共表達時二聚體形成丟失。標本用抗AGR2、抗FLAG和鏈霉親和素免疫印跡。在輸入樣品中使用微管蛋白作為加載對照。代表了兩個獨立的實驗。F競爭IP表明,隨著AGR2共表達的增加,二聚體的形成隨濃度的增加而喪失。標本用抗AGR2、抗FLAG和鏈霉親和素免疫印跡。在輸入樣品中使用微管蛋白作為加載對照。代表了兩個獨立的實驗。

圖5. AGR2中催化死亡的C81S和致病的H117Y突變使其失去了結合和抑制IRE1β活性的能力。A圖,在PyMol中顯示了AGR2(PDB:2LNS)的結構,并指出了相關的突變。紫色和灰色的卡通描繪了兩個AGR2分子及其二聚體結構38,特定的殘基用球棒表示。B使用AGR2突變體的競爭IP示意圖概述。C競爭IP顯示C81S和H117Y AGR2突變體對二聚體的抑制作用喪失。用抗agr2、抗FLAG-IRE1β和鏈霉親和素免疫印跡標本。在輸入樣品中使用微管蛋白作為加載對照。代表了兩個獨立的實驗。D CALU-1ERN1-/-IRE1βFLAG-DOX細胞經構建的β基因(野生型或突變體)導入后,經1μg/ml多西環素誘導72小時后,IRE1AGR2FLAG-DOX過表達。對轉基因表達72小時后D細胞系的細胞死亡進行量化。培養皿中所有細胞經Annexin V和Live/Dead染色后進行流式細胞儀分析。所有單陽性細胞和雙陽性細胞均為死亡細胞。代表兩個獨立的實驗,有兩個重復。
在RT-qPCR實驗中,I.DOT非接觸式納升級移液系統用于將cDNA、引物、SYBR混合液精準轉移至384孔板,每孔反應體積均一(納升級),避免交叉污染,顯著提升定量準確性與實驗通量,為IRE1β活性相關基因表達的高通量驗證提供技術支撐(對應Figure 1E-F、3A、3F)
非杯狀細胞(Calu-1)中過表達IRE1β會引發劇烈細胞死亡與XBP1剪接(Figure 1C-E),而杯狀細胞樣LS174T中IRE1β活性顯著降低(Figure 1C、1E)。AP-MS與Co-IP驗證顯示,AGR2與IRE1β特異性結合,不與IRE1α互作,且體內小鼠結腸組織中也存在該相互作用(Figure 2B-E),提示AGR2是IRE1β的杯狀細胞特異性調控因子。
Calu-1細胞中共表達AGR2可顯著降低IRE1β介導的XBP1剪接與RIDD活性(Figure 3A),并完全逆轉其細胞毒性(Figure 3B-C);反之,LS174T細胞中siRNA敲低AGR2會增強IRE1β活性(Figure 3E-G),且IRE1β抑制劑4μ8C可逆轉該效應,證明AGR2直接抑制IRE1β功能。
凝膠過濾層析顯示,AGR2共表達會使IRE1β的二聚體峰向單體峰偏移(Figure 4B-C);競爭Co-IP實驗表明,AGR2劑量依賴性破壞IRE1β-Avi與IRE1β-FLAG的二聚體形成(Figure 4E-F),直接證明AGR2通過阻止IRE1β二聚化使其失活。
AGR2的催化死突變C81S(喪失二硫鍵異構酶活性)與IBD相關突變H117Y,均無法與IRE1β結合,也不能破壞其dimer或抑制細胞毒性(Figure 5C-E);而單體突變體(E60A、K64A)仍保留調控功能,說明AGR2的催化結構域與H117位點是結合IRE1β的關鍵,與二聚化能力無關。
本研究的核心突破在于揭示了AGR2作為IRE1β的“分子變阻器”,通過杯狀細胞特異性表達精準調控IRE1β的核酸酶活性,進而平衡黏液折疊負荷與內質網(ER)穩態。這一發現的核心價值體現在三方面:從機制創新來看,首次證實AGR2可通過直接結合IRE1β的柔性環區域,破壞其二聚體形成,從而抑制IRE1β的XBP1剪接與RIDD活性,填補了IRE1β特異性調控機制的空白,且與BIP調控IRE1α的模式形成鮮明的旁系同源物(paralogue)特異性調控網絡;從疾病關聯來看,炎癥性腸病(IBD)相關的AGR2H117Y突變會導致其喪失對IRE1β的調控功能,引發IRE1β過度激活,這可能是杯狀細胞功能異常與腸道炎癥的關鍵誘因,為IBD精準治療提供了全新靶點;從技術支撐來看,I.DOT非接觸式納升級移液系統在RT-qPCR中的應用,保障了384孔板高通量基因定量的準確性與重復性,尤其是納升級體積的精準分配,不僅減少了樣品損耗與交叉污染,還為分子機制的快速驗證(如XBP1剪接效率、靶基因表達定量)提供了高效工具,助力高通量篩選與機制驗證的無縫銜接。
未來研究可進一步深入探索AGR2與IRE1β的結合動力學特征,以及黏液折疊負荷如何通過調控二者相互作用影響IRE1β活性;同時,基于本研究揭示的調控機制,開發靶向AGR2-IRE1β軸的小分子藥物,有望為IBD等黏液屏障相關疾病提供新的治療策略。此外,I.DOT在本研究中的成功應用,也為類似分子機制研究提供了可復用的技術范式,將進一步推動未折疊蛋白反應(UPR)通路調控網絡的系統解析,為理解更多分泌細胞的穩態調控機制奠定基礎。

同騰睿杰(上海)生物技術有限公司作為CYTENA I.DOT中國總代理商,為您提供優質的售前售后服務。
聯系電話:021-50826962
聯系郵箱:sales@ttbiotech.com
1、Tschurtschenthaler, M. et al. Defective ATG16L1-mediated removal of IRE1alpha drives Crohn's disease-like ileitis. The Journal of experimental medicine 214, 401-422.
2、Tsuru, A. et al. Negative feedback by IRE1beta optimizes mucin production in goblet cells. Proceedings of the National Academy of Sciences of the United States of America 110, 2864-2869.
3、Al-Shaibi, A. A. et al. Human AGR2 Deficiency Causes Mucus Barrier Dysfunction and Infantile Inflammatory Bowel Disease. Cellular and molecular gastroenterology and hepatology 12, 1809-1830.
4、Bertoli-Avella, A. et al. A disorder clinically resembling cystic fibrosis caused by biallelic variants in the AGR2 gene. Journal of medical genetics 59, 993-1001.

